1. Poisson-Boltzmann, PB模型
============================
在量子化学计算和使用经典分子力学进行模拟中,常会用到隐式溶剂模型,特别是作为一种快速估算溶剂化自由能的方法,在蛋白-底物分子作用计算,和蛋白质折叠等方面特别有用。量子化学gauss中,还会经常用来估算溶剂中某些分子的pKa。那么,隐式溶剂如何做到不使用溶剂而估算溶剂的影响呢?它的原理是什么?
我们首先从最基本的问题谈起,即溶剂化效应是怎么回事。溶剂化效应是发生在溶剂中的很多化学现象的关键。它影响反应的自由能势能面,改变反应速率和热力学平衡。我们知道,一旦知道一个反应的势能面,我们就知道了它的全部热力学和动力学信息。举个例子,比如乙烷的二面角的旋转,如果我们把二面角作为反应坐标,则如果我们知道了这个旋转势能面,那么我们就知道了每个二面角取值的能量(势能),那么根据Boltzmann分布,我们可以计算任意温度下,乙烷对各二面角取值的分布概率:
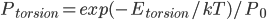
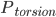 是能量为
是能量为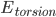 的构象的权重,而
的构象的权重,而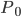 是配分函数,是把所有构象的权重加起来。
是配分函数,是把所有构象的权重加起来。
这个概率就是所谓的热力学平衡分布,即体系平衡后某构象的浓度等于总浓度乘以该构象的概率:

而动力学,就是翻越构象能垒的速率,可由能垒高度ΔE得到:
反应速率常数: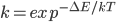
反应速率由阿伦尼乌斯公式,即产物浓度变化: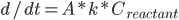 (
(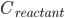 为反应物浓度)
为反应物浓度)
由上面可知反应势能面真的很厉害。有了反应势能面,别无他求。有些初学者往往觉得跑个Gauss没什么,做MD才给力。其实,如果量子化学计算能够计算势能面,干嘛要跑MD呢?MD不过是模拟体系在那个势能面上随时间的变化而已。不过,量子化学计算个单分子在气相中的反应,搜个过渡态啥的,很准确。但是对于溶剂中的反应呢?这时候,溶剂的作用,即溶剂分子在反应物分子周围来来往往的影响就不能不考虑了。回到上面乙烷的例子,如果在水里面旋转,肯定要受到水分子的阻力什么的影响。那么多的水分子,随便哪个分子动一下,阻力就不一样,这就是熵效应,即所有水分子的不同构象对乙烷旋转势能面的影响。在考虑了周围溶剂的影响后,乙烷的势能面已经不叫势能面了,而叫旋转自由能势能面。自由能包括内能和熵两个部分。当然,熵不仅包括溶剂的,也包括乙烷自身的构象熵,比如它自身在水里面的振动,转动,平动,都影响最后的自由能势能面结果。
那么,怎么考虑溶剂化效应对反应的势能面的影响呢?即,如果我们要计算自由能势能面,那有2个方法:第一,跑MD,随机模拟溶液体系的一些构象,然后根据体系中乙烷取各构象的概率,反推出自由能势能面;第二,不跑MD,还是用量子化学的方法来计算单个乙烷的旋转势能面,但是使用隐式溶剂模型,即使用一个经验的公式,来把溶剂的影响的平均结果考虑进来,这样,直接得出自由能势能面。
怎么平均化溶剂作用影响呢?学过高中物理的都知道,设置一个等效介电常数。即,真空中两电荷作用力 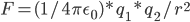
那,放到溶剂中,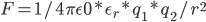
比如,水的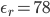
当然,我们在拉格朗日图景中,要使用能量而不是牛顿力学中的力。即,我们要求的双点电荷的Coulomb势能U(r12):
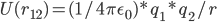
十分简单。But,wait a minute...实际的隐式溶剂模型,并不使用Coulomb公式,而是使用Poisson-Boltzmann公式!!!
为什么呢? 模型不一样。Coulomb公式用的是点电荷模型,q1, q2,距离r。但是,对于真实的分子,是由原子核加核外电子云构成,电子云是有体积的。量子化学计算就是核外电子云的密度变化。即使是使用经典分子力学力场, AMBER, CHARMM什么的,也是把原子看成是带电荷的,有半径r的球的,这样,必然每个原子附近都有不同的电荷密度。如果把原子看作点电荷,进行对加和计算,则收敛慢且截断误差很大。一个更聪明,更快速的办法是:把体系分成均匀格点,计算每个格点的静电势Φ(r),然后在该点上的电荷q(r)所具有的能量则为
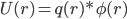
这样,最后把各点上的U(r)加起来,就得到总的静电势能U(注意静电势与静电势能的差别)。这就是Poisson方法的思想。Poisson与Coulomb对加和计算的差别,类似于量子化学计算中DFT方法与HF的差别, PB把Coulomb的多体作用转化为随空间变化的密度问题,势能等于密度乘以静电势。现在,最关键的,是静电势如何得到?它是由Poisson解决的。简单说,Poisson给出了一个微分方程,这个微分方程描述了介电常数,电荷密度分布和静电势的关系,给出了一个由一大片电子云或几个空间分布的带电原子球ρ(r)计算静电势Φ(r)的关系。
Poisson公式:
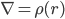
∇:拉普拉斯算符,可理解为一阶偏导
r: 表示空间格点,你可以把空间用线切割开,把r想象成一个个格点位置(注意跟上面的表示两电荷距离的r定义不同),格点是使用有限差分方法求解微分方程的常用方法
ε: 介电常数
Φ:所要求的静电势
ρ:各格点上的电荷密度分布
这里,Φ(r)是代表体系r处的静电势,是我们要求的量, 它的二阶导由该点的电荷密度ρ(r)及其介电常数ε(r)决定。虽然这个公式的物理意义我理解不了,但是我只需要知道通过它我们可以得到静电势Φ(r)就足够了,即由上式可得:
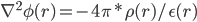
这样,我们得到在各点r处静电势能的二阶导∇^2Φ(r),最后对整个盒子所有格点进行二重数值积分即可得到最终静电势,乘以各点上的电荷得到静电势能U(r) = q(r)*Φ(r)。MD中一般使用的计算静电势能的PME方法,即Particle Mesh Ewald方法,原理就是利用Poisson公式进行格点积分。
上面只说了Poisson公式,不是说隐式溶剂用的是Poisson-Boltamann公式吗?Boltzmann干了啥?
问题是这样的,上面的模型,用来计算蛋白质在一定浓度电解质溶液中的静电势能,存在一个系统误差,原因是没有考虑反离子的分布的贡献。蛋白质往往是带电荷的,结果在真实溶液体系中,在一个蛋白的不同表面处,往往分布着较高浓度的相反电荷。这叫离子氛模型。它造成的结果是,溶液中的静电作用的衰减要比由库仑公式计算的势能面的衰减快得多。这叫离子屏蔽效应。电解质溶液理论中使用Debye长度来屏蔽效应的强弱。但是Debye长度是一个经验量,无法很方便地定义。怎么能考虑这个屏蔽效应呢?
一个直接的想法是,把溶质比如蛋白附近的反离子浓度计算出来。即一个改变了的电荷分布: ρ(r)+ Δρ(r),其中后面一项Δρ(r)就是溶液体系中反离子的浓度在r处的分布。
即: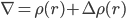
通过这个改进的Poisson公式,可以更好的考虑蛋白质在电解质溶液即离子溶液中的静电势能。当然,由于反离子分布本身改变静电势能,而静电势能又改变反离子浓度分布,因此,这必然是个自洽计算。
那么怎么计算反离子浓度呢? Poisson-Boltzmann公式中的Boltzmann部分:
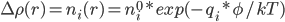
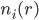 : 溶液中离子种类i在r处的数密度(可以是分数个)
: 溶液中离子种类i在r处的数密度(可以是分数个)
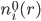 : 溶液中离子种类i的平均数密度
: 溶液中离子种类i的平均数密度
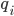 : 该种类离子的电荷
: 该种类离子的电荷
 :盒子空间中该区域的静电势
:盒子空间中该区域的静电势
上式的物理意义是,某离子在盒子内某处数密度对平均密度的偏差,与其在该处的造成的静电势能的大小成自然幂反比关系。换句话说,在蛋白附近处静电势Φ越高,则该处的反离子浓度越高,同号离子的浓度越低。因为前者造成能量下降,后者造成能量上升。
对于一般尺寸的盒子,由于在数千到数万个格点上进行含指数计算的自洽计算,收敛相当相当慢,伤不起,只好对该指数进行tylor展开为多项式,如果截取多项式到第一项,则ni与Φ为一个线性关系,这样的近似称为线性PB计算LPBE. LPBE对于不带净电荷的小分子来说是足够准确的;但是,对于带电荷的,特别是电荷浓度较大的体系,通常要取到第二项,ni是Φ的三次方的函数,这样的近似称为非线性PB计算NPBE。这方面的论文很多,尝试用各种妖娆的数值方式加快计算。简单了解一下上面这些名词很有必要。
在很多采用一阶近似的线性PB模型中,为了避免屏蔽效应考虑的不充分,会在Boltzmann那一部分的前面加上一个系数 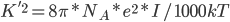 , 直接考虑Debye-Hukel效应。其中
, 直接考虑Debye-Hukel效应。其中 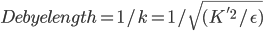
2. Gernalized Born - GB模型
===========================
为了计算一个半径为r,带净电荷为q的球的溶剂化自由能,即把它从气相移到介电常数为ε的溶剂中,Born提出了一个及其简单的计算公式:
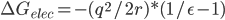
可以简单理解为,如果你想往溶剂里移动了一个电荷q,那么溶剂必然会被极化,在电荷所在位置产生了一个同号的q,你的移动必须克服这电荷与镜像之间的排斥力。
这个模型的奇妙之处在于只有一个可调参数,即半径r. 一开始born使用原子的共价半径r,计算结果虽然误差较大,但定性准确。考虑到模型的简单性,可以说是出乎意料。后来有人用vdW半径计算,误差大大减小,但仍需提高。于是,不断有人试图使用不同的半径,经验关联的,量子化学计算的。总而言之,Born半径是Born模型的关键。
Generalize Born模型,即广义Born模型,或一般化了的Born模型,是用它来估算分子的溶剂化自由能,即把分子中的每个组成原子用上面的模型估算一下,然后加和得到总的溶剂化自由能;你会问,分子表面的原子与溶剂的作用比较强,分子内部的原子与溶剂几乎没有作用,怎么能加和呢?不要紧,给分子不同位置的原子设置不同的有效半径即可,这个有效半径可以用来反应原子离表面的距离,或者说被埋的程度。另外,你必须注意到,每个原子不仅与自身的镜像作用,而且还会与其它原子作用。
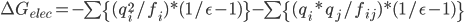
前面一项就是原来的Born,后面一项是原子间两两作用。这两项中的r都被f取代了。f的计算:
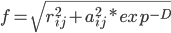 f等于原子间距rij和原子born半径aij按指数衰减的均方根;
f等于原子间距rij和原子born半径aij按指数衰减的均方根;
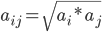 其中有效半径aij等于两原子的有效半径的几何平均;
其中有效半径aij等于两原子的有效半径的几何平均;
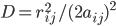 D是f中aij^2按指数衰减的幂,这个衰减类似于考虑周围反离子分布的屏蔽效应;
D是f中aij^2按指数衰减的幂,这个衰减类似于考虑周围反离子分布的屏蔽效应;
GB模型是PB模型的特例。(To be addressed)
当然,这里仅仅计算了静电作用对溶剂化自由能的贡献。还有一部分贡献来自于分子体积破坏了水分子间的氢键网络,从而产生的能量代价。这一部分的贡献对于中性分子的溶剂化自由能大概占10%。怎么计算这一部分的能量呢?先问破坏了H键网络的后果是什么呢?水分子在分子表面形成了表面张力,由朗格谬而,表面形成能等于表面张力乘以表面积,所以这部分自由能必然是表面积的线性函数。所以,要计算的就是溶剂可及表面solvent accessible area, SA。由于一般使用vdw半径作为探测溶剂可及表面的基准,所以可称之为ΔGvdw。
即: ΔGvdw = σ*A
最终: 
因此,这种估算溶剂化自由能的方法称为GB/SA模型,分子模拟中有时称为MM/GBSA。必须注意有报道称MM/GBSA模拟多肽的构象得到的结果与显式溶剂模型的结果有显著的不同(参考英文wiki: implicit solvation - GBSA条目),其MM/GBSA不能正确识别蛋白native的结构。还有盐桥过于稳定的毛病,可能是因为屏蔽效应估计不足;alpha-helix的分布也明显高估。看来,这个PB的近似模型尽管简单,但是要慎重使用阿。
要想提高计算精度,可以使用PB/SA模型,即静电部分的贡献,占溶剂化自由能的主要部分(>90%),使用PB来计算。当然,这一部分要在几千个格点上做自洽计算,所以要慢的多。
http://emuch.net/html/201112/3965375.html