![]()
![]()
![]()
Next: Glutathione Up: Topology Tutorial Previous: Getting Started
Examining the Topology File
Consider the diatomic hydrogen molecule, H![]() . In molecular dynamics simulations, this molecule is modeled as two point masses connected by a simple spring. In this respect, two sets of information are needed to simulate the H
. In molecular dynamics simulations, this molecule is modeled as two point masses connected by a simple spring. In this respect, two sets of information are needed to simulate the H![]() molecule:
molecule:
- atomic connectivity information (to tell the MD program that one atom is bonded to the other)
- the spring stiffness and equilibrium (bond) length
The above simple example demonstrates that if you wish to simulate a more complicated molecule, the MD force field will come in two parts:
- topology file (top_all27_prot_lipid.inp)
- parameter file (par_all27_prot_lipid.inp)
The topology file defines which atoms are connected to one another through chemical bonds. Different atomic topologies are shown in Figure 2. The topology file may specify ``bonds" (2 atoms connected), ``angles" (3 atoms connected), and ``dihedrals" (4 atoms connected linearly). If you have a complete list of 2-atom bonds, the angle and dihedral information may be inferred from it. Thus, angle and dihedral information is extraneous and may be omitted. Given a complete atom list and set of bonds, psfgen is able to construct the correct topology for the molecule. However, ``improper" angles, which are used to restrain chiral and planar centers must be specified explicitly. The improper interaction is used to keep the amino acid peptide bond planar.
![\begin{figure}\begin{center}
\includegraphics[height=2.4in]{pictures/bondstretch-red}\end{center}\end{figure}](http://www.ks.uiuc.edu/Training/Tutorials/science/topology/topology-html/img10.gif) |
The parameter file quantifies the variables which are used in the force field potential energy. It gives parameters such as the stiffness and equilibrium value of an angle between 3 atoms, etc.
Today, we are going to use the topology and parameter files listed above: top_all27_prot_lipid.inp and par_all27_prot_lipid.inp. These files contain the CHARMM22 protein and the CHARMM27 lipid topologies and parameters.
The CHARMM force field contains topology and parameter information for the standard 20 amino acids, lipids, nucleic acids, and some other organic molecules. Thus, one can simulate any protein, DNA, or molecular systems which are composed of these basic ``building blocks". But what if you wanted to simulate a system that contained a molecule not described in the CHARMM topology and parameter files? At first glance, it might seem like you need to build an entire set of new parameters for your molecule, a process which may involve computationally expensive quantum chemistry calculations. However, this might not be the case.
There are a wide variety of natural and non-natural amino acids and other molecules that can be simulated by piecing together parameters already in the CHARMM parameter set. You are restricted only by the bond, angle, improper and dihedral angles already available for your atom types. If you wish to study a protein system involving non-natural amino acids, it may be possible to run simulations using existing parameters for similar standard amino acids. In this manner, you will be saved much effort in not being forced to develop new parameters for your system.
L- and D- Amino Acids
Imagine you have a peptide composed of both L- and D-amino acids. Cyclosporin, an immunosuppressant drug, for example, contains D-alanine. D-alanine is also used in bacterial cell wall synthesis. There are currently 46 files in the Protein Data Bank which contain D-alanine (Example, 1ICO). You might wish to simulate the molecule, but the standard CHARMM22 force field contains only L-amino acids. However, since the molecules are identical in all respects except for their chirality, the parameters for D-amino acids are identical to their L-configured counterparts. Figure 3 shows the chiral difference between both molecules.
![\begin{figure}\begin{center}
\includegraphics[height=1.0in]{pictures/LD-alanine}\end{center}\end{figure}](http://www.ks.uiuc.edu/Training/Tutorials/science/topology/topology-html/img11.gif) |
We will develop the new topology file for D-alanine using the existing topology file for L-alanine.
- Unix/Mac OS X Users: In a terminal window go to your 1-examine directory and open the topology file top_all27_prot_lipid.inp with your text editor:
cd 1-examine nedit top_all27_prot_lipid.inp - Windows Users: Navigate to your 1-examine directory using Windows Explorer and open the topology file top_all27_prot_lipid.inp with your text editor by right-clicking the file, selecting the Open With... option and choosing the program WordPad.
- 1
- Find the topology entry for alanine using the Search or Find feature in your text editor by typing ``ala" in the search field.
- 2
- The entry for alanine is shown on the next page.
[frame=single, framerule=1.2mm, framesep=3mm, label=Alanine Topology Entry, fontsize=\scriptsize] RESI ALA 0.00 GROUP ATOM N NH1 -0.47 ! | ATOM HN H 0.31 ! HN-N ATOM CA CT1 0.07 ! | HB1 ATOM HA HB 0.09 ! | / GROUP ! HA-CA--CB-HB2 ATOM CB CT3 -0.27 ! | \ ATOM HB1 HA 0.09 ! | HB3 ATOM HB2 HA 0.09 ! O=C ATOM HB3 HA 0.09 ! | GROUP ! ATOM C C 0.51 ATOM O O -0.51 BOND CB CA N HN N CA BOND C CA C +N CA HA CB HB1 CB HB2 CB HB3 DOUBLE O C IMPR N -C CA HN C CA +N O DONOR HN N ACCEPTOR O C IC -C CA *N HN 1.3551 126.4900 180.0000 115.4200 0.9996 IC -C N CA C 1.3551 126.4900 180.0000 114.4400 1.5390 IC N CA C +N 1.4592 114.4400 180.0000 116.8400 1.3558 IC +N CA *C O 1.3558 116.8400 180.0000 122.5200 1.2297 IC CA C +N +CA 1.5390 116.8400 180.0000 126.7700 1.4613 IC N C *CA CB 1.4592 114.4400 123.2300 111.0900 1.5461 IC N C *CA HA 1.4592 114.4400 -120.4500 106.3900 1.0840 IC C CA CB HB1 1.5390 111.0900 177.2500 109.6000 1.1109 IC HB1 CA *CB HB2 1.1109 109.6000 119.1300 111.0500 1.1119 IC HB1 CA *CB HB3 1.1109 109.6000 -119.5800 111.6100 1.1114
The entries in the topology file provide the following information:
- 3
- RESI ALA 0.00indicates a new residue with the name ALA and total charge of 0.00.
- 4
- GROUPindicates that the following atoms (up to the next GROUP command) are part of a ``group" of atoms which carries an integer charge. Groups are often made up of atoms which interact electronically, sharing electron density.
- 5
- ATOM N NH1 -0.47 ! |indicates a new atom with the name N, the type NH1, and a charge of -0.47. The ! sign is a comment signal. Anything appearing after it on the line will be ignored. Thus, the stick representation of alanine is simply a comment in the file. It is only there so the user may see the topology of the residue. Note that atoms N, HN, CA, and HA form a group with net charge 0.00.
- 6
- BOND CB CA N HN N CAindicates sets of 2 atoms which are connected by a single bond. The bonds are created between consecutive atoms. Thus, one bond is placed between atoms CB and CA, one is placed between N and HN, and one between N and CA. The BOND entries on the next line are read exactly the same way, and are put on a new line simply for readability.
- 7
- DOUBLE O Csimply indicates that the atoms named O and C are bonded. DOUBLE is a synonym for BOND and will not affect the resulting psf file. The fact that there is a double bond between the atoms is accounted for in the parameters, not in the topology.
- 8
- IMPR N -C CA HN C CA +N Oindicates sets of 4 atoms for which the improper bond interaction will be calculated. The chiral center is listed first. An improper bond is created between atoms N, C, CA, and HN, with N being at the center, and one is created between C, CA, N, and O with C at the center. Recall, improper bonds are created to maintain planarity, and these two maintain the planarity of the protein backbone.
- 9
- DONOR and ACCEPTOR linesare not used by the CHARMM force field or NAMD. They indicate which atoms may act as hydrogen bond donors and acceptors, but since the CHARMM force field no longer includes an explicit hydrogen bonding energy term, these entries are ignored. Note that any hydrogen bonds you observe in your simulations using the latest CHARMM force field are purely the result of electrostatic interactions.
- 10
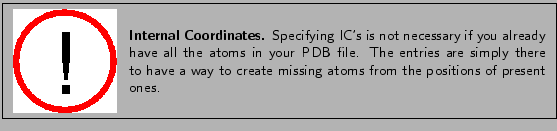
- IC -C CA *N HN 1.3551 126.4900 180.0000 115.4200 0.9996IC stands for ``internal coordinates". They provide a complete set of coordinates for atoms relative to other atoms in the residue. Thus, if atoms are missing from a structure (such as H atoms from an x-ray crystallography PDB file), the complete molecule may still be built based on the positions of existing atoms. The IC records define the bond length, angle, dihedral (or improper) angle for groups of 4 atoms. The pattern may come in 2 forms:
- IC A B C D [bond(AB)] [angle(ABC)] [dihedral(ABCD)] [angle(BCD)] [bond(CD)]
- IC A B *C D [bond(AC)] [angle(BCA)] [improper(ABCD)] [angle(BCD)] [bond(CD)]. The * next to the C atom indicates that it is at the center of an improper angle definition. No * indicates that the 4 atoms do not have an improper topology.
In the example above, atoms C and N have a bond length of 1.3551 Å, atoms CA, N, and C have an angle of 126.4900
 between them, atoms C, CA, N, and HN have a 180.0000 improper angle, atoms CA, N, and HN have an angle of 115.4200 between them, and atoms N and HN have a bond length of 0.9996 Å. Atom N is the central atom in the improper topology.
between them, atoms C, CA, N, and HN have a 180.0000 improper angle, atoms CA, N, and HN have an angle of 115.4200 between them, and atoms N and HN have a bond length of 0.9996 Å. Atom N is the central atom in the improper topology.
Now, how can we use the above topology entry for L-alanine to create a topology for D-alanine?
- 11
- Go to your text editor and copy the existing entry for alanine. Use the mouse to highlight every alanine entry line (from RESI ALA 0.00 to IC HB1 CA *CB HB3 1.1109 109.6000 -119.5800 111.6100 1.1114). Then click Edit
 Copy.
Copy.
- 12
- Paste the copied entry below the original alanine entry by using the Edit Paste option.
- 13
- Edit your copied alanine entry to make it D-alanine by changing the following lines:
Change:
to:
Change:
to:
Change:
to:RESI ALA 0.00
RESI DAL 0.00
IC N C *CA CB 1.4592 114.4400 123.2300 111.0900 1.5461
IC N C *CA CB 1.4592 114.4400 -123.2300 111.0900 1.5461
IC N C *CA HA 1.4592 114.4400 -120.4500 106.3900 1.0840
IC N C *CA HA 1.4592 114.4400 120.4500 106.3900 1.0840
The only difference between the two molecules is the inverse chirality of the methyl side chain and HA atom about the CA, or ![]() -carbon, atom. Thus, by changing 123.23 to -123.23 in the first IC, we change the L-chiral center at the CA atom to a D- chiral centre. The same principle applies to the second IC command.
-carbon, atom. Thus, by changing 123.23 to -123.23 in the first IC, we change the L-chiral center at the CA atom to a D- chiral centre. The same principle applies to the second IC command.
- 14
- Click File Save As..., enter top_all27_prot_lipid_d-ala.inp as your new filename, and click Enter. You have now created a topology file with D-alanine included!
Windows Users: Make sure you save the file in .txt format.

Lysine and Ornithine
Next, let's consider a natural amino acid that is not normally incorporated into proteins. Ornithine is similar to lysine but is one carbon atom shorter in its side-chain:
Lysine:
|
HN-N
| HB1 HG1 HD1 HE1 HZ1
| | | | | /
HA-CA--CB--CG--CD--CE--NZ--HZ2
| | | | | \
| HB2 HG2 HD2 HE2 HZ3
O=C
|
Ornithine:
|
HN-N
| HB1 HG1 HD1 HE1
| | | | /
HA-CA--CB--CG--CD--NE--HE2
| | | | \
| HB2 HG2 HD2 HE3
O=C
|

Let's say that you want to probe the effect of side-chain length of a particular lysine residue within your protein. You could run a simulation incorporating ornithine into the protein. However, a topology entry would be needed for the residue. Furthermore, if existing atom types were suitable to be used in the ornithine molecule, you would not need to create parameters. In this section, we will demonstrate how to create a topology entry for ornithine.
Construction of a topology entry for ornithine (or any new molecule) from existing topology entries involves assigning atom types and charges based on similar molecules. Therefore, it is useful to sketch out the new molecule in order to determine which parts are similar to other molecules for which topology and parameter information already exists. Since ornithine is very similar to lysine, we have sketched lysine below and isolated its groups. Then, we have matched up similar groups in ornithine.
![\begin{figure}\begin{center}
\includegraphics[height=1.2in]{pictures/lys-orn-step1}\end{center}\end{figure}](http://www.ks.uiuc.edu/Training/Tutorials/science/topology/topology-html/img17.gif) |
As you can see, the above case is rather simple. An ornithine topology entry may be created from lysine by simply removing one of lysine's aliphatic carbon atoms, along with the hydrogen atoms bonded to it. Diagrams such as the one above are very useful when constructing a topology entry.
Now that we know which lysine groups will be used to construct the ornithine topology entry, what exact changes are needed to make it? We will begin by examining the lysine topology entry
- 1
- In your 1-examine directory, open your D-alanine topology file with your text editor by:
- Unix/Mac OS X Users: typing the following in your terminal window:
nedit top_all27_prot_lipid_d-ala.inp - Windows Users: right-clicking the file top_all27_prot_lipid_d-ala.inp, selecting the Open With... option and choosing the program WordPad.
- Unix/Mac OS X Users: typing the following in your terminal window:
- 2
- Find the topology entry for lysine using the Search or Find feature in your text editor by typing ``lys" in the search field.
- 3
- The entry for lysine is shown on the next page.
[frame=single, framerule=1.2mm, framesep=3mm, label=Lysine Topology Entry, fontsize=\scriptsize] RESI LYS 1.00 GROUP ATOM N NH1 -0.47 ! | ATOM HN H 0.31 ! HN-N ATOM CA CT1 0.07 ! | HB1 HG1 HD1 HE1 HZ1 ATOM HA HB 0.09 ! | | | | | / GROUP ! HA-CA--CB--CG--CD--CE--NZ--HZ2 ATOM CB CT2 -0.18 ! | | | | | \ ATOM HB1 HA 0.09 ! | HB2 HG2 HD2 HE2 HZ3 ATOM HB2 HA 0.09 ! O=C GROUP ! | ATOM CG CT2 -0.18 ATOM HG1 HA 0.09 ATOM HG2 HA 0.09 GROUP ATOM CD CT2 -0.18 ATOM HD1 HA 0.09 ATOM HD2 HA 0.09 GROUP ATOM CE CT2 0.21 ATOM HE1 HA 0.05 ATOM HE2 HA 0.05 ATOM NZ NH3 -0.30 ATOM HZ1 HC 0.33 ATOM HZ2 HC 0.33 ATOM HZ3 HC 0.33 GROUP ATOM C C 0.51 ATOM O O -0.51 BOND CB CA CG CB CD CG CE CD NZ CE BOND N HN N CA C CA BOND C +N CA HA CB HB1 CB HB2 CG HG1 BOND CG HG2 CD HD1 CD HD2 CE HE1 CE HE2 DOUBLE O C BOND NZ HZ1 NZ HZ2 NZ HZ3 IMPR N -C CA HN C CA +N O DONOR HN N DONOR HZ1 NZ DONOR HZ2 NZ DONOR HZ3 NZ ACCEPTOR O C IC -C CA *N HN 1.3482 123.5700 180.0000 115.1100 0.9988 IC -C N CA C 1.3482 123.5700 180.0000 107.2900 1.5187 IC N CA C +N 1.4504 107.2900 180.0000 117.2700 1.3478 IC +N CA *C O 1.3478 117.2700 180.0000 120.7900 1.2277 IC CA C +N +CA 1.5187 117.2700 180.0000 124.9100 1.4487 IC N C *CA CB 1.4504 107.2900 122.2300 111.3600 1.5568 IC N C *CA HA 1.4504 107.2900 -116.8800 107.3600 1.0833 IC N CA CB CG 1.4504 111.4700 180.0000 115.7600 1.5435 IC CG CA *CB HB1 1.5435 115.7600 120.9000 107.1100 1.1146 IC CG CA *CB HB2 1.5435 115.7600 -124.4800 108.9900 1.1131 IC CA CB CG CD 1.5568 115.7600 180.0000 113.2800 1.5397 IC CD CB *CG HG1 1.5397 113.2800 120.7400 109.1000 1.1138 IC CD CB *CG HG2 1.5397 113.2800 -122.3400 108.9900 1.1143 IC CB CG CD CE 1.5435 113.2800 180.0000 112.3300 1.5350 IC CE CG *CD HD1 1.5350 112.3300 122.2500 108.4100 1.1141 IC CE CG *CD HD2 1.5350 112.3300 -121.5900 108.1300 1.1146 IC CG CD CE NZ 1.5397 112.3300 180.0000 110.4600 1.4604 IC NZ CD *CE HE1 1.4604 110.4600 119.9100 110.5100 1.1128 IC NZ CD *CE HE2 1.4604 110.4600 -120.0200 110.5700 1.1123 IC CD CE NZ HZ1 1.5350 110.4600 179.9200 110.0200 1.0404 IC HZ1 CE *NZ HZ2 1.0404 110.0200 120.2700 109.5000 1.0402 IC HZ1 CE *NZ HZ3 1.0404 110.0200 -120.1300 109.4000 1.0401
- 4
- Copy the existing lysine entry. Use the mouse to highlight every lysine entry line (from RESI LYS 1.00 to IC HZ1 CE *NZ HZ3 1.0404 110.0200 -120.1300 109.4000 1.0401). Then click Edit Copy.
- 5
- Paste the copied entry below the original lysine entry by using the Edit Paste option.
You will edit your copied lysine entry to make it an ornithine entry by making the following changes:
- 6
-
Change:
to:RESI LYS 1.00
RESI ORN 1.00
- 7
- Delete the appropriate carbon atom and hydrogen atoms. We will delete the
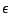 -carbon (CE) atom and the hydrogens bonded to it (HE1 and HE2), but we must be careful! In deleting the -carbon, we are deleting atoms which are part of a group with NZ. Thus, the
-carbon (CE) atom and the hydrogens bonded to it (HE1 and HE2), but we must be careful! In deleting the -carbon, we are deleting atoms which are part of a group with NZ. Thus, the 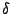 -carbon and its hydrogens will become part of this group.
-carbon and its hydrogens will become part of this group.
- Delete the following lines:
GROUP ATOM CD CT2 -0.18 ATOM HD1 HA 0.09 ATOM HD2 HA 0.09 - Change the following lines:
Change:
to:
Change:
to:
Change:
to:
Change:
to:
Change:
to:
Change:
to:
Change:
to:ATOM CE CT2 0.21
ATOM CD CT2 0.21
ATOM HE1 HA 0.05
ATOM HD1 HA 0.05
ATOM HE2 HA 0.05
ATOM HD2 HA 0.05
ATOM NZ NH3 -0.30
ATOM NE NH3 -0.30
ATOM HZ1 HC 0.33
ATOM HE1 HC 0.33
ATOM HZ2 HC 0.33
ATOM HE2 HC 0.33
ATOM HZ3 HC 0.33
ATOM HE3 HC 0.33
- Delete the following lines:
- 8
- In considering bonding, we must delete the bonds between CE and all other atoms, and take care to rename the NZ and HZ atoms appropriately.
Change:
to:
Change:
to:
Change:
to:BOND CB CA CG CB CD CG CE CD NZ CE
BOND CB CA CG CB CD CG NE CD
BOND CG HG2 CD HD1 CD HD2 CE HE1 CE HE2
BOND CG HG2 CD HD1 CD HD2
BOND NZ HZ1 NZ HZ2 NZ HZ3
BOND NE HE1 NE HE2 NE HE3
- 9
- Even though the hydrogen bond DONOR terms are unused, we still change them to maintain consistency in atom names.
Change:
to:
Change:
to:
Change:
to:DONOR HZ1 NZ
DONOR HE1 NE
DONOR HZ2 NZ
DONOR HE2 NE
DONOR HZ3 NZ
DONOR HE3 NE
- 10
- Since we delete 3 atoms from the lysine molecule in order to create ornithine, 3 IC commands become obsolete.Delete the following lines:
IC CB CG CD CE 1.5435 113.2800 180.0000 112.3300 1.5350 IC CE CG *CD HD1 1.5350 112.3300 122.2500 108.4100 1.1141 IC CE CG *CD HD2 1.5350 112.3300 -121.5900 108.1300 1.1146
- 11
- You need to be very careful when renaming atoms and updating the ICs accordingly. For our ornithine molecule change the atom names in the following lines. The atomic arrangements (bond lengths, angles, etc.) stay the same, since we simply removed one carbon atom from the side chain.
Change:
to:
Change:
to:
Change:
to:
Change:
to:
Change:
to:
Change:
to:IC CG CD CE NZ 1.5397 112.3300 180.0000 110.4600 1.4604
IC CB CG CD NE 1.5397 112.3300 180.0000 110.4600 1.4604
IC NZ CD *CE HE1 1.4604 110.4600 119.9100 110.5100 1.1128
IC NE CG *CD HD1 1.4604 110.4600 119.9100 110.5100 1.1128
IC NZ CD *CE HE2 1.4604 110.4600 -120.0200 110.5700 1.1123
IC NE CG *CD HD2 1.4604 110.4600 -120.0200 110.5700 1.1123
IC CD CE NZ HZ1 1.5350 110.4600 179.9200 110.0200 1.0404
IC CG CD NE HE1 1.5350 110.4600 179.9200 110.0200 1.0404
IC HZ1 CE *NZ HZ2 1.0404 110.0200 120.2700 109.5000 1.0402
IC HE1 CD *NE HE2 1.0404 110.0200 120.2700 109.5000 1.0402
IC HZ1 CE *NZ HZ3 1.0404 110.0200 -120.1300 109.4000 1.0401
IC HE1 CD *NE HE3 1.0404 110.0200 -120.1300 109.4000 1.0401

After making all the corrections, your topology definition for ornithine should look as shown on the next page.
[frame=single, framerule=1.2mm, framesep=3mm, label=Ornithine Topology Entry, fontsize=\scriptsize] RESI ORN 1.00 GROUP ATOM N NH1 -0.47 ! | ATOM HN H 0.31 ! HN-N ATOM CA CT1 0.07 ! | HB1 HG1 HD1 HE1 ATOM HA HB 0.09 ! | | | | / GROUP ! HA-CA--CB--CG--CD--NE--HE2 ATOM CB CT2 -0.18 ! | | | | \ ATOM HB1 HA 0.09 ! | HB2 HG2 HD2 HE3 ATOM HB2 HA 0.09 ! O=C GROUP ! | ATOM CG CT2 -0.18 ATOM HG1 HA 0.09 ATOM HG2 HA 0.09 GROUP ATOM CD CT2 0.21 ATOM HD1 HA 0.05 ATOM HD2 HA 0.05 ATOM NE NH3 -0.30 ATOM HE1 HC 0.33 ATOM HE2 HC 0.33 ATOM HE3 HC 0.33 GROUP ATOM C C 0.51 ATOM O O -0.51 BOND CB CA CG CB CD CG NE CD BOND N HN N CA C CA BOND C +N CA HA CB HB1 CB HB2 CG HG1 BOND CG HG2 CD HD1 CD HD2 DOUBLE O C BOND NE HE1 NE HE2 NE HE3 IMPR N -C CA HN C CA +N O DONOR HN N DONOR HE1 NE DONOR HE2 NE DONOR HE3 NE ACCEPTOR O C IC -C CA *N HN 1.3482 123.5700 180.0000 115.1100 0.9988 IC -C N CA C 1.3482 123.5700 180.0000 107.2900 1.5187 IC N CA C +N 1.4504 107.2900 180.0000 117.2700 1.3478 IC +N CA *C O 1.3478 117.2700 180.0000 120.7900 1.2277 IC CA C +N +CA 1.5187 117.2700 180.0000 124.9100 1.4487 IC N C *CA CB 1.4504 107.2900 122.2300 111.3600 1.5568 IC N C *CA HA 1.4504 107.2900 -116.8800 107.3600 1.0833 IC N CA CB CG 1.4504 111.4700 180.0000 115.7600 1.5435 IC CG CA *CB HB1 1.5435 115.7600 120.9000 107.1100 1.1146 IC CG CA *CB HB2 1.5435 115.7600 -124.4800 108.9900 1.1131 IC CA CB CG CD 1.5568 115.7600 180.0000 113.2800 1.5397 IC CD CB *CG HG1 1.5397 113.2800 120.7400 109.1000 1.1138 IC CD CB *CG HG2 1.5397 113.2800 -122.3400 108.9900 1.1143 IC CB CG CD NE 1.5397 112.3300 180.0000 110.4600 1.4604 IC NE CG *CD HD1 1.4604 110.4600 119.9100 110.5100 1.1128 IC NE CG *CD HD2 1.4604 110.4600 -120.0200 110.5700 1.1123 IC CG CD NE HE1 1.5350 110.4600 179.9200 110.0200 1.0404 IC HE1 CD *NE HE2 1.0404 110.0200 120.2700 109.5000 1.0402 IC HE1 CD *NE HE3 1.0404 110.0200 -120.1300 109.4000 1.0401
- 12
- Click File Save As..., enter top_all27_prot_lipid_orn.inp as your new filename, and click Enter. You have now created a topology file with ornithine (and D-alanine) included.
Windows Users: Make sure you save the file in .txt format.
![\begin{figure}\begin{center}
\includegraphics[height=1.3in]{pictures/lys-orn-step2}\end{center}\end{figure}](http://www.ks.uiuc.edu/Training/Tutorials/science/topology/topology-html/img21.gif) |
![\framebox[\textwidth]{
\begin{minipage}{.2\textwidth}
\includegraphics[width=2...
... entries. A solution is presented at the end of the tutorial.}
\end{minipage} }](http://www.ks.uiuc.edu/Training/Tutorials/science/topology/topology-html/img22.gif)
![\begin{figure}\begin{center}
\includegraphics[height=1.0in]{pictures/nle-step0}\end{center}\end{figure}](http://www.ks.uiuc.edu/Training/Tutorials/science/topology/topology-html/img23.gif)
4-Hydroxyproline
In this section we will try to create the topology entry of another non-standard amino acid by again looking up its components in the corresponding topology entries for these known components. While this method has worked well up to this point, it must be realized that it is not foolproof, and may not work in more complicated situations.
4-Hydroxyproline is a non-standard amino acid involved in post-translational modification. The hydroxylation occurs via an unusual enzyme named proline hydroxylase.
4-Hydroxyproline:
HD1 HD2
| \ /
N---CD HG
| \ /
| CG
| / \
HA-CA--CB OE--HE
| / \
| HB1 HB2
O=C
|
Proline:
HD1 HD2
| \ /
N---CD HG1
| \ /
| CG
| / \
HA-CA--CB HG2
| / \
| HB1 HB2
O=C
|
Although it may not seem to be a very big change in the chemical formula, the replacement of H by OH is not possible to treat with the standard topology library at our disposal.
As you may have already noticed, all the five atoms on the ring of proline have unique atom types. For example, the atom type of the nitrogen atom on proline ring is N which is defined as ``proline N", instead of NH1 which is the normal atom type for peptide nitrogen. The atom with name CG is of type CP2, again, unique to proline. Thus, bonding an O atom to CG would require choosing a type for the O atom, which is impossible, since there is no topology entry with O bonded to a proline or proline-like ring. We simply do not have parameters for oxygen bonded to a 5-member proline ring.
As a result, we would have to re-parameterize this new amino acid if we wished to work with it. New atom types and parameters would be created from quantum chemistry calculations in order to properly model the new ring.
In this section you will learn how to apply the lessons in Unit 1 from beginning to end, building a new topology file, using it to generate a psf file for the system, and running a new simulation with the topology you created.
![]()
![]()
![]()
Next: Glutathione Up: Topology Tutorial Previous: Getting Started