Exploring the landscape of large scale conformational changes such as protein folding at atomic detail poses a considerable computational challenge. Coarse-grained representations of the peptide chain have therefore been developed(1).Coarse-grained representations combined with enhanced computer power allow the simulation of system of biologically relevant size(submicrometric) and time scale(us or ms)(2).
The beads model is a model based on a united-atom representation of the amino acid(involving one to six interaction centers).

<1> Elastic network models
The system is represented by a network of beads connected by elastic springs,usually one bead per amino acid.
<2> 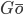 -like models
-like models
The model was propsed specifically for the simulation of protein folding. In the original version, the protein is represented as a chain of one-bead amino acids whose structure is biased toward the native configuration by means of simple attractive and repulsive non-bonded interaction between beads.
In the model, amino acids are represented by point particles, or beads, which are located at the positions of the Ca atoms. Consecutive beads in the chain are tethered by an anharmonic potential: 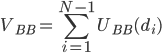 , where
, where 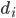 is the distance between beads i and i+1,
is the distance between beads i and i+1,
The use of an advanced sampling method like replica exchange can be used to rapidly explore conformational space.
Both ENM and model depend on reference configuration which makes them weakly transferable to general dynamics studies.
References:
1. Insights from Coarse-Grained Go Models for Protein Folding and Dynamics.
2.Coarse-grained models for proteins