Biological processes in the human body are dependent on highly specific molecular interactions. The vast majority
of the interactions take place in compartments within the cell, and an understanding of the behavior of the
membranes that compartmentalize and enclose the cell is therefore critical for rationalizing these processes. Biological
membranes are complex structures formed mostly by lipids and proteins. For this reason lipid bilayers
have received a lot of attention both computationally and experimentally for many years.[amber 2015 manual,p43]

reference:
Fliposomes: pH-Sensitive Liposomes Containing a trans-2-morpholinocyclohexanol-Based Lipid That Performs a Conformational Flip and Triggers an Instant Cargo Release in Acidic Medium
Here is a link of lipid library developled by CHARMM-GUI.
The objective of this project is to simulate the dynamics of liposome in different pH, hopefully to find out the mechanism of conformation change of liposome upon protonation. In this tutorial, I will be using one liposome MorC16 as an example to show the procedure of the modeling and simulation.

Molecular Modelling
create molecule by gaussian viewer,and then optimize the molecule:
%chk=morc16.chk
%Nproc=26
# opt am1 geom=connectivity
save the molecule as morc16.mol2
One thing I want to check is the position of morpholino ring.
# opt=modredundant pm3 geom=connectivity
add at the end of gjf file
D 3 4 27 15 S 36 10.0
[blank]
the results of scan:

so there are two positions correspond to two energy minima, which are loan pair of N face -OH, or face back to -OH:

According to the assumption, the hydrogen bond should form between N and hydroxyl O, and the distance is 3.64 which is fit for hydrogen bond. I saved this structure as morc16_head1.mol2.
copy morc16_head1.mol2 to morc16.mol2, and submit to Swissparam.
retrieve morc16.zip after about one minute,
unzip the file and get: morc16.crd/itp/mol2(charge added)/par/pdb/psf/rtf.
convpdb.pl -segnames -out charmm22 morc16.pdb |sed "s/LIG/MOR/g"|sed "s/PRO0/LIG/g">tmp.pdb
mv tmp.pdb morc16.pdb
genPSF.pl -par param=22 morc16.pdb >morc16_charmm.psf
genPSF.pl -xplor -par param=22 morc16.pdb >morc16_xplor.psf
In topology file , the charges are poorly defined, we use charge information from gaussian log file to update the charges in topology file. This time we run the gaussian optimization with:
%chk=morc16.chk
%Nproc=26
# opt(if you don't want optimization, delete this) b3lyp/6-31g(d) pop=nbo geom=connectivity
assign_charge.pl -nbo -log morc16.log -rtf ./toppar/morc16.rtf
To facilitate future simulation, we add topology and parameters to top_all22_prot_cmap_phmd.inp and par_all22_prot_cmap_phmd.inp, and replace the default topology and parameter files in system(/home/zwei/softwares/c37a2/toppar/par_all22_prot.inp:top_all22_prot.inp).
add "PATCHING FIRS NONE LAST NONE" at the end of topology definition
let's run an equilibration simulation of morc16 monomer to find a equilibrated structure.
vmd morc16_head1.mol2, open molefacture, set up segname, residue name,chain,residue id,and save as morc16.pdb
minCHARMM.pl -par param=22 morc16.pdb >tmp.pdb
mdCHARMM.pl -par param=22,dynsteps=50000 -trajout morc16_equi_va.dcd -final morc16_equi_va.pdb tmp.pdb
| VA | GB |
From the equilibration we observed the monomer is likely to keep two tails together in vacuum, and try to separate from each other in implicit solvent.
Protonated patch for MOR:
add a hydrogen to morc16_head1.mol2,
%chk=morc16_h.chk
%Nproc=26
# opt b3lyp/6-31g(d) pop=nbo geom=connectivity
change charge from 0 1 to 1 1
cp ./toppar/morc16.rtf ./toppar/morc16_h.rtf
edit morc16_h.rtf, add ATOM HD HNR 0.0000 to the end of ATOM list, then run:
assign_charge.pl -nbo -log morc16_h.log -rtf ./toppar/tmp.rtf(check sum of charges, sometime it is not integer!!!)
edit ~/softwares/c37a2/toppar/top_all22_prot.inp
create PRES MOR1 1.000
copy GROUP and the rest from tmp.rtf to top_all22_prot.inp
and add
BOND N HD
IC HD N C4 C3 1.03 105.02 34.99 111.61 1.53
pH simulation of monomer
we use vacuum equilibrated structure to build bilayer
copy morc16_equi_va.pdb morc16.pdb
copy add_h.inp to ./MORC16/
make sure the name as in red are "mor" and "morp",
"if ?selresn eq mor patch morp(or whatever patch you defined mor1,mor2...) @segid @ires"
then run:
charmm in=morc16 <add_h.inp
If the warning of "No improper parameters for..." showed up, you need to add those parameters to par_all22_prot_cmap_phmd.inp.
output: morc16_h.pdb/psf/crd
copy water.crd to ./toppar/
copy solvate.inp to ./MORC16/
charmm in=morc16 <solvate.inp(change Xleng or Yleng or Zleng if you want to)
outputs:morc16_h_waterbox.crd/str/prm/pdb
morc16_h_solvated.psf/pdb/str/crd
copy add_ions_hphmd.inp to ./MORC16/
charmm in=morc16 conc=0.100<add_ions_hphmd.inp
outputs:morc16_h_ions.pdb/str/prm/crd
copy minimize.inp to ./MORC16/
charmm in=morc16 <minimize.inp
outputs:morc16_h.solv.ion.min.psf/pdb/crd/pbc
the system should be looks like this:

Now we use this system to find parameter A and B for this molecule.
copy phmd.in radius_gbsw.str to ./toppar/
edit phmd.in
MOR 5.0 0.0 0.0 0.0
ATOM [protonated charge] [unprotonated charge]
......
HD [protonated charge] [unprotonated charge] 1.0 0.0
copy hphmd_derive.inp to ./MORC16/
charmm <hphmd_derive.inp
extract theta and dE/dtheta value from lamb file and fit the data by xgrace.

Replace A=-580.696 b=0.735614 barrier=2.0
For constant pH simulation, copy hphmd.inp to ./ , set up pH value, and run the simulation:
charmm <hphmd.inp
For replica exchange simulation, copy hphmd_phrex.inp to ./, set up pH conditions, and run:
mpirun -np # charmm <hphmd_phrex.inp
outputs:morc16.ph#.dcd/ene/lamb/rst
The lambda file format is:
#ititr 1 (running number for titation degrees of freedom)
#ires 1 (titration group corresponding to residue number in PDB file)
#itauto 0(single site LYS/ARG/ASPP/GLUPP,0;two-site, one pKa ASPP2/GLUP2,3 4; two-site,different pKa HSP,1 2)
#Park 4.118 ( ln(10)RT(pH-pKa^mod))
step number ,lambda(lambda =0 for protonated, 1 for unprotonated) Obtain unprotonated fractions from CPHMD simulation CptSX.pl lambda file first step last step pH.(here step means frame, from 1 to ....) output files: flipsome.min_h.ph7.0.lamb-1-54.sx # ires pH unprot pure mixed 1 1 7.0 0.00 0.84 0.16 2 2 7.0 0.00 0.86 0.14 ..... 96 96 7.0 0.00 0.90 0.10 Calculate pKa values by fitting to Henderson-Hasselbalch (HH) equation cptpKa,pl flipsome.min_h flipsome.min_h.ph7.0.lamb-1-54.sx output file: flipsome.min_h.ph7.0.lamb-1-54.pka 1 NAN 2 NAN 3 NAN 4 NAN 5 NAN 6 NAN 7 NAN 8 NAN 9 NAN 10 NAN 11 NAN 12 NAN 13 NAN 14 NAN 15 7.58 1.0 16 NAN ..... 96 NAN
The fliposome illustrated here is Eth_C16: 
First thing we need to deal with is the modelling the lipid, including build the structure, topology, parameters for force field, then we run the constant pH simulation with charmm(maybe gromacs in future), we will show these step by step follow a tutorial that can be found at the bottom of this page.
A. Modelling of the lipid system
We use gaussian viewer to create this molecule, saved as mol2 format(ethc16.mol2), topology and parameter files were created by SwissParam (http://www.swissparam.ch/).  After we submit the structure, we will get a zip file which include *.crd(coordinate file);
After we submit the structure, we will get a zip file which include *.crd(coordinate file);
*.itp(topology file for gromacs);
*.par(parameter file for charmm);
*.pdb,*.psf;*.rtf(topology file for charmm)
mkdir data under ETHC16, and copy ethc16.pdb and ethc16.psf to ./data In VMD, open tcl/tk console, Before we create bilyer, align the molecule to principal axes, and make the long tail aligned to z-axis(charmm can also do this).
package require Orient
namespace import Orient::orient
set sel [atomselect top "all"]
set I [draw principalaxes $sel]
set A [orient $sel [lindex $I 2] {0 0 1}]
$sel move $A
set I [draw principalaxes $sel]
set A [orient $sel [lindex $I 1] {0 1 0}]
$sel move $A
set I [draw principalaxes $sel]
then save the aligned structure,$sel write pdb ethc16.pdb
then we will make copies from this file
source lipid.tcl (change the number when making the serials of residues, charmm require the residue number to be unique some time.) lipid -l *(name of *) -x (xsize) -y (ysize) example: lipid -l ethc16 -x 20 -y 20 then the bilayer will be created:  Next is to edit titratable residue and add hydrogen to the lipids. In lipid.rtf, we add a patch to residue LIG: PRESI LIGP 1.000 GROUP ATOM N NR 0.20 ! | ATOM H8 HNR 0.36 ! HD--N--H8 ATOM HD HNR 0.44 ! | BOND N HD ANGLE HD N C2 DIHE HD N C2 C1 HD N C2 C3 DONOR HD N IC HD N C2 C1 0.00 0.00 120.00 0.00 0.00 then run the srcript: charmm in=lipid <add_h.inp the output files include: lipid_h.pdb lipid_h.psf lipid_h.crd
Next is to edit titratable residue and add hydrogen to the lipids. In lipid.rtf, we add a patch to residue LIG: PRESI LIGP 1.000 GROUP ATOM N NR 0.20 ! | ATOM H8 HNR 0.36 ! HD--N--H8 ATOM HD HNR 0.44 ! | BOND N HD ANGLE HD N C2 DIHE HD N C2 C1 HD N C2 C3 DONOR HD N IC HD N C2 C1 0.00 0.00 120.00 0.00 0.00 then run the srcript: charmm in=lipid <add_h.inp the output files include: lipid_h.pdb lipid_h.psf lipid_h.crd  Now the system is ready to be used to run simulation in implicit solvent model, if you want explicit solvent model, run: charmm in=lipid <solvate.inp the outputs are: *_h_solvated.crd, *_h_solvated.psf, *_h_solvated.pdb, *_h_solvated.str, *_h_waterbox.crd,*_h__waterbox.pdb,*_h_waterbox.str,*_h_waterbox.prm Now we add ions to the system charmm in=lipid conc=0.100 <add_ions_hphmd.inp then we minimize the system: charmm in=lipid <minimize.inp the system should be look like this
Now the system is ready to be used to run simulation in implicit solvent model, if you want explicit solvent model, run: charmm in=lipid <solvate.inp the outputs are: *_h_solvated.crd, *_h_solvated.psf, *_h_solvated.pdb, *_h_solvated.str, *_h_waterbox.crd,*_h__waterbox.pdb,*_h_waterbox.str,*_h_waterbox.prm Now we add ions to the system charmm in=lipid conc=0.100 <add_ions_hphmd.inp then we minimize the system: charmm in=lipid <minimize.inp the system should be look like this 
B. Running PH simulations
We use PHMD Module in charmm to simulate the dynamics of titrable groups under specific pH condition. lambda = 1 for deprotonated states and lambda=0 for protonated states. Lambda itself is function of theta which can freely propagate without need for restrictions, when theta = 0, +/- 2n(PI), lambda = 0. When theta = pi+/- 2n(PI), then lambda = 1. The sin^2 function also provides a natural double well for quadratic energy functions of lambda. Add the contents below to then end of lipid_phmd.in LIG 9.0 9523809 -0.00027 2.0 N 0.20 -0.36 H8 0.36 0.36 HD 0.44 0.00 1.00 0.00 8.5 is the experimental value from NMR with methanol, 9.7 is predicted from ACD/LABS, so I put 9.0 as experimental value for water solvent. Before we run the constant pH simulation, we need to derive parameters for the input file, which most importantly A and B. According to charmm documents:http://www.charmm.org/documentation/c34b1/phmd.html PHMD PAR 23 wri 25 PH @ph NPRI @prfrq PHFRQ 1 mass 1.E30 BARR 0 - DERI BETA 5.0 TEMP 300.0000 LAM PHTEST num 1 set 0.4(theta from 0.4 to 1.4) I only run 0.001ps, the manual suggests 1ns. the trajectory output : step theta dE/dtheta CPHMD simulation, we run: charmm <hphmd.inp lipid.ph9.dcd .ene .lamb .rst will be generated under ./data pH replica exchange simulation, we run: mpirun -np 8 charmm<hphmd_phrex.inp(pH from 1 to 15, delta pH 2) Now we run NAMD simulation to equilibrate this system: namd2 +p 8 flipsome.namd (GBIS)
We also run the equilibration with charmm: minCHARMM.pl -par xtop=lipid.rtf,xpar=lipid.par flipsome.pdb >flipsome.min.pdb mdCHARMM.pl -par gb,xtop=lipid.rtf,xpar=lipid.par,dynsteps=10000 -trajout flipsome_charmm.dcd -log flipsome.log flipsome.min.pdb(minimize it before equilibration):
Now the bilipid system is ready, we run CPHMD simulation by: charmm < hphmd.inp > hphmd.out & clear lld output: flipsome.min_h.dcd flipsome.min_h.ene flipsome.min_h.rst flipsome.min_h.ph7.0.lamb. The lambda file format is: #ititr 1 2 3 ....... 96(running number for titation degrees of freedom) #ires 1 2 3 ...... 96(titration group corresponding to residue number in PDB file) #itauto K 0 0 0.....0(single site LYS/ARG/ASPP/GLUPP,0;two-site, one pKa ASPP2/GLUP2,3 4; two-site,different pKa HSP,1 2) #Park 4.667 4.667 4.667 .......4.667 ( ln(10)RT(pH-pKa^mod)) step number ,lambda(lambda =0 for protonated, 1 for unprotonated) 10 0.19 0.20 0.19..........0.30 20 0.11 0.06 0.16 .........0.00 ........ 500 0.00 0.00 0.00........0.00 Obtain unprotonated fractions from CPHMD simulation CptSX.pl lambda file first step last step pH.(here step means frame, from 1 to ....) output files: flipsome.min_h.ph7.0.lamb-1-54.sx # ires pH unprot pure mixed 1 1 7.0 0.00 0.84 0.16 2 2 7.0 0.00 0.86 0.14 ..... 96 96 7.0 0.00 0.90 0.10 Calculate pKa values by fitting to Henderson-Hasselbalch (HH) equation 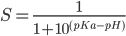 cptpKa,pl flipsome.min_h flipsome.min_h.ph7.0.lamb-1-54.sx output file: flipsome.min_h.ph7.0.lamb-1-54.pka 1 NAN 2 NAN 3 NAN 4 NAN 5 NAN 6 NAN 7 NAN 8 NAN 9 NAN 10 NAN 11 NAN 12 NAN 13 NAN 14 NAN 15 7.58 1.0 16 NAN ..... 96 NAN
cptpKa,pl flipsome.min_h flipsome.min_h.ph7.0.lamb-1-54.sx output file: flipsome.min_h.ph7.0.lamb-1-54.pka 1 NAN 2 NAN 3 NAN 4 NAN 5 NAN 6 NAN 7 NAN 8 NAN 9 NAN 10 NAN 11 NAN 12 NAN 13 NAN 14 NAN 15 7.58 1.0 16 NAN ..... 96 NAN
Solvate the protein
To be continued...... This is a link of lipid builder developed by: the Laboratory for Molecular Modeling/EPFL http://lipidbuilder.epfl.ch/home CHARMM-GUI provides a web-based graphical user interface to generate various molecular simulation systems and input files to facilitate and standardize the usage of common and advanced simulation techniques. Currently, CHARMM-GUI supports CHARMM, NAMD, GROMACS, and OpenMM simulation programs mostly based on the CHARMM force fields. http://www.charmm-gui.org/