Theory of protein titration curves. General equations for impenetrable spheres--Charles Tanford and John G. Kirkwood,1957
Before this paper, the theory of titration curves of impenetrable protein had been based on a model which represents the protein molecules as a sphere with a continuous and uniform distribution of charge on its surface. This paper replace it with a more realistic one which the charges are taken to be discrete uinit charges located at fixed positions.
The titration curve itself is a plot of the average number of bound protons, 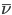 versus pH.
versus pH.
Protein dipoles langevin dipoles ---Warshel &Russell, 1984 ,1985
adiabtic charging approach based on umbrella sampling method---Warshel &Sussman
Notably, Warshel and coworkers were the first to employ MD methods to improve calculation of pKa values in proteins.
pKa’s of Ionizable Groups in Proteins: Atomic Detail from a Continuum Electrostatic Model
---Donald Bashford and Martin Karplus 1990
Simulation of protein conformational freedom as a function of pH:constant pH molecular dynamics using implicit titration.
---Baptista AM,1997
Potential of mean force is used to account for an implicit titration.
Simulating proteins at constant pH An approach combining molecular dynamics and monte carlo simulation---Roland Burgi,2002
The current methods to analyze the titration behavior of ionisable sites depend on determing the free energy difference of protonating the site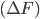 and relating it to a pKa value through the relation.
and relating it to a pKa value through the relation.
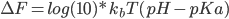
The free energies of protonating or deprotonating a residue are calculated through MD simulation,whereas MC steps sample the protonating states of the ionisable residues during an MD simulation.
CPHMD ---Constant pH molecular dynamics using continuous titration coordiantes 2004
Microscopic description of protein and macroscopic description of solvent(GB). This method employ an extended Hamiltonian in which continuous titration coordinates propagate simultaneously with the atomic motions of the system.
In the last decade, simple continuum models had been developed to determine the pKa shifts in proteins. The general idea in these calculations is to use an implicit water model, such as continuum electrostatic theory or Langevin dipole water model to estimate the pKa shifts of a static structure due to eletrostatic forces in the protein environment. These calculation is based on a single static X-ray structure, this paper average the pKa shifts over multiple conformations. One disadvantage of simple continuum model is solute dielectric must be manually selected since it essentially determine the magnitude of the pKa shifts. They also mention use LRA to address configurational averaging issue.
coupling of conformational dynamics to titration events refers to weighting protein configurations based on the energetic favorability of certain protonation states.
Toward the accurate first-principles predcition of ionization equilibria in proteins 2006(The pKa calculation based on CPHMD, they improved generalized born implicit solvent model with approxiate Debye-Huckel screening function to account for salt effects and REX for enhanced conformational sampling.)